Molecular dynamics of carbon and classical and quantum hydrogen in iron
In the HEmS project, we got interested in how hydrogen behaves in the crystal lattice of α-iron. Part of our approach was to make molecular dynamics (MD) simulations, and on this page I want to show off some movies that we made.
In order to do MD you need a way to calculate interatomic forces. In all our work we use the quantum mechanical magnetic tight binding approximation [1]. Details of the models can be found in Ref [2].
First we made some classical MD simulations to look at the dynamics of hydrogen, H, and carbon, C, when they are trapped at a vacancy (that is a lattice site with an atom missing). These two movies show simulations at 400K.
The left hand, or upper if your display is small, movie (simulation time, 2.9 picoseconds) has two C atoms and one H atom trapped in the vacancy. Note these points:
The two carbon atoms form a "dimer" at such an angle in the cube that outlines the vacancy so that each C atom has its preferred four bonds—one to the other C and three of equal length and tetrahedral bond angles to Fe atoms (see Ref. [2]).
The H atom is trapped in the vacant site, it cannot escape in reasonable time. However it clearly avoids the C atoms. They are happily bonded and there is no motivation to form C–H bonds (although I have heard metallurgists talking of hydrocarbons being formed within steel).
The right hand, or lower, movie (simulation time, 3.6 picoseconds) shows two hydrogen atoms trapped at the vacancy. We know that the vacancy will trap up to five H atoms and that they like to occupy positions close to the centres of the faces of the cube enclosing the vacancy [3].
Our movie shows that, although I started them off on adjacent faces, they don't like that. They soon migrate to opposite faces; and you'll see if you follow the movie that even if one of them moves to an adjacent face the other moves to the opposite face to avoid it. Authors who invent so called classical potentials for hydrogen in iron (for example Song and Curtin) talk of the need for H–H terms in the physics, but this is nonsense. What is keeping them apart is not "H–H repulsion", but the elastic strains that they are imposing on the iron crystal lattice.
You can see that although they have the opportunity to do so, they do not form a hydrogen molecule. This is because they are living in the electron gas of the transition metal and if they were to get close enough to form a bond, it would have zero bond energy since both bonding and antibonding level would be filled. The H cannot escape as its escape rate is very low compared to the simulation time here [4].
Hydrogen is the lightest element and generally speaking it is not sufficient to treat is as a classical object—you would never treat electrons as classical, which is why we use quantum mechanics to describe interatomic forces. In the first two videos it's almost legitimate to use classical MD since the temperature is 400K; but even at room temperature hydrogen behaves non classically—its diffusivity as a function of temperature is non-Arrhenius as it can tunnel between tetrahedral sites of the perfect iron crystal lattice.
If I showed a video of hydrogen in perfect iron at room temperature then it would not last long as the H atom would quickly wander away out of shot (it's not trapped as in the vacancy movies above). So the next movie (simulation time 5 picoseconds) shows hydrogen in perfect iron at 100K. It does jump between tetrahedral sites but no too frequently in the total simulation time of 5 picoseconds.
We do not treat the quantum mechanics of the electrons and the H nucleus—the proton—on the same footing. The electrons are represented as Bloch states of the tight binding hamiltonian; the proton dynamics are simulated using path integral molecular dynamics, based on Richard Feynman's path integral representation of the quantum partition function. In this theory, a quantum particle is simulated as a "necklace" of beads, each bead is an image of the proton. The dynamics of this object is represented in the movie exactly as such a necklace. Even the iron atoms look a bit fuzzy as their nuclei are treated in the same way, and indeed in that picture their position is indeed uncertain as expressed in Heisenberg's uncertainty principle.
Note that these simulations are made in a smaller simulation cell that the 128 atoms of the first movies, above. Here the view shows two atomic cubes—the one on the lower left is behind that on the upper right. It's important to get this perspective when we come to look at the next movie (simulation time 50 picoseconds).
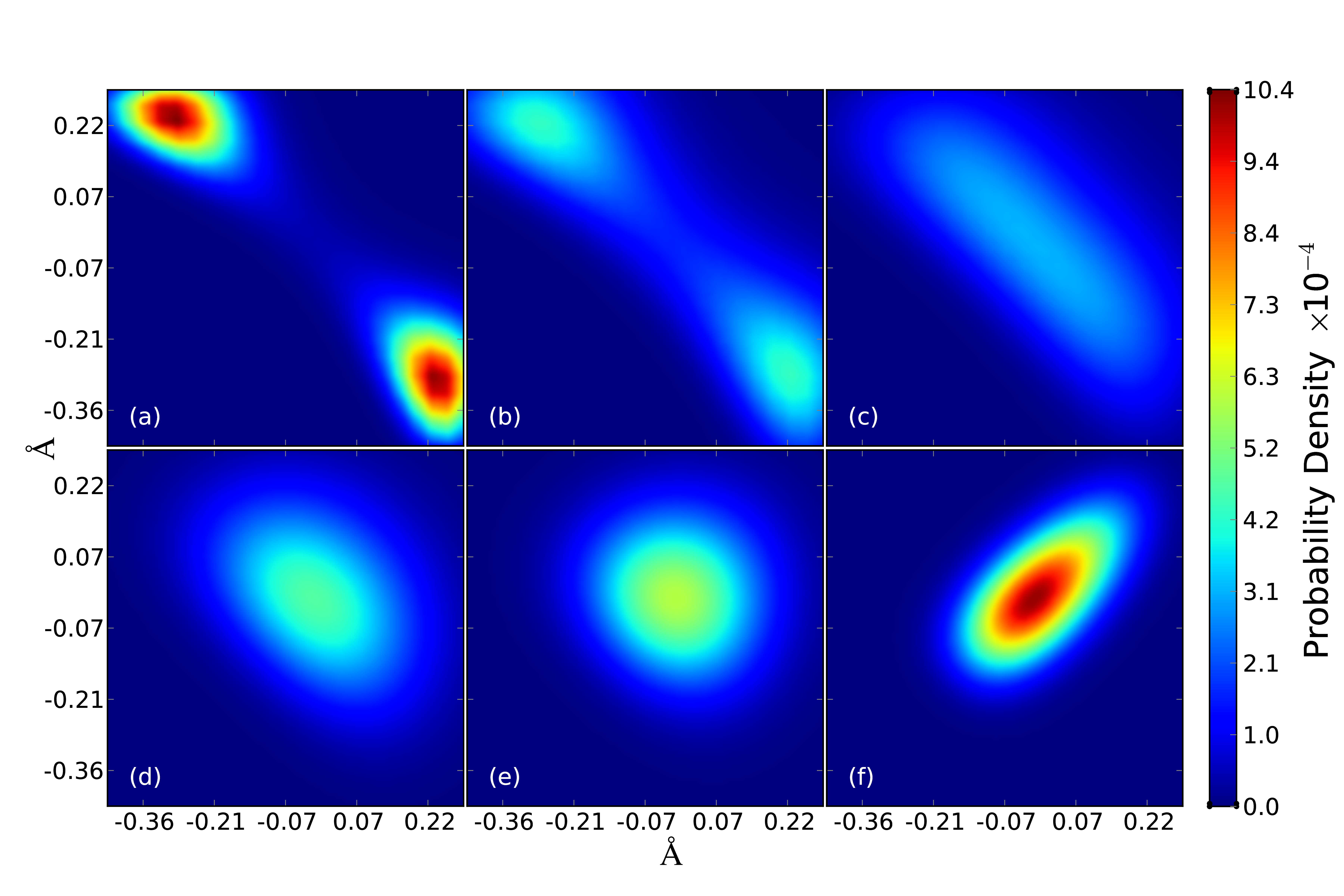
The delocalised nature of the proton is evident from the spread of the beads in the necklace. At high temperature the "springs" connecting the beads stiffen and the necklace shrinks to a (classical) point. If you follow the movie closely and for long enough you will see brief glimpses of the proton delocalised between two tetrahedral sites: it has "split into two"! See the figure next to the movie; this shows the particle position probability density calculated from the path integral partition function in the case that the centroid of the particle is held at the saddle point between two stable perfect tetrahedral sites in the iron lattice [5]. The temperatures in the figure are (a) T = 20 K; (b) T = 50 K; (c) T = 100 K; (d) T = 200 K; (e) T = 300 K; and (f) T = 1000 K.
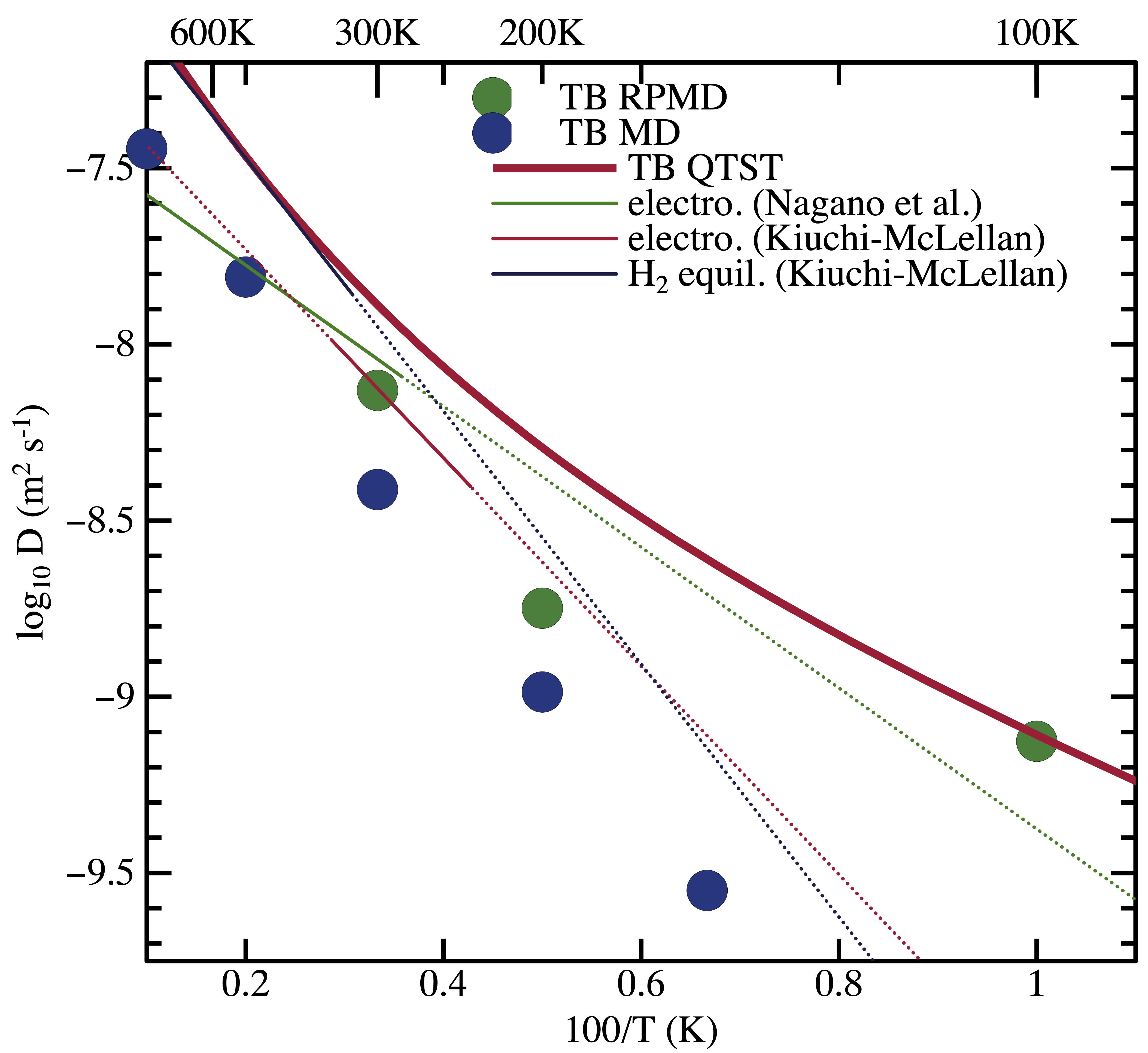
The right hand (or lower) figure shows the diffusivity of H in Fe calculated using both classical and path integral molecular dynamics (RPMD). You can see the non Arrhenius behaviour at low temperature that I mentioned earlier. You can also see comparisons between the explicity MD calculations and the diffusivity calculated using the quantum transition state theory [4]. This is the first time that these have been compared using the same hamiltonian [4,5].
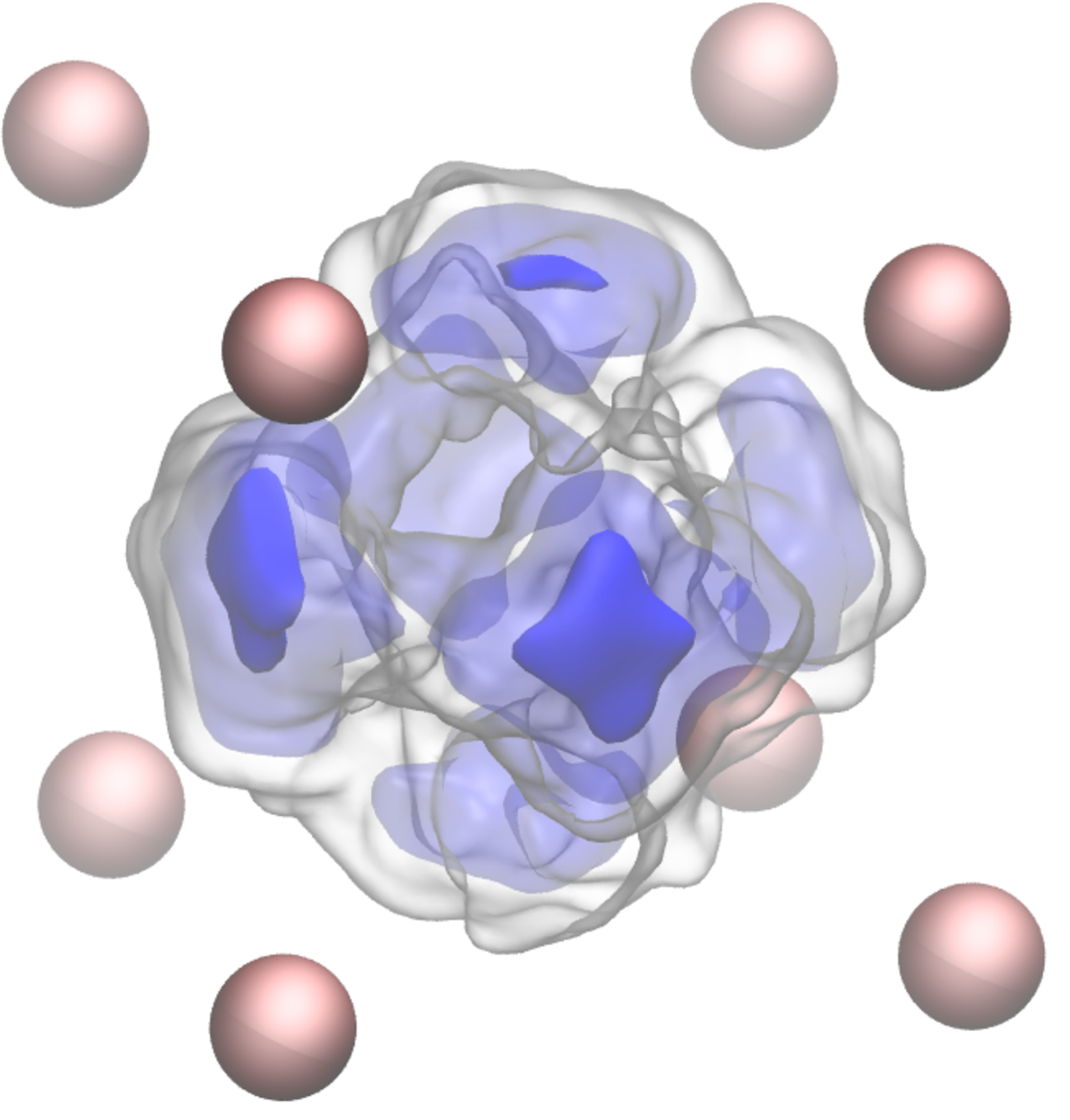
To show that the proton is quite noticeably delocalised even at room temperature, the final movie shows the proton, again trapped at a vacancy so that it won't wander away. Note the perspective is the same as before and now the cube in front has its body centred atom missing.
Note that, just as in the classical simulation above, the proton occupies the sides of the cube bounding the vacant site. The hydrogen atom does not occupy the site left vacant by the Fe atom as would a substitutional impurity. Interstitials in steel—C, N, H— do not substitute on the host lattice.
This is illustrated in the right hand figure, taken from ref [6], which is the particle probability density for the hydrogen atom trapped at the vacancy. You can see that the proton is delocalised. This means that it acquires quantum kinetic energy and that its energy landscape is highly anharmonic. We found in ref [6] that these nuclear quantum effects contribute to as much as 25% of the energy to trap the proton at a vacant site. These effects are of course a part of the measured trap energy but they are not captured in the usual quantum mechanical calculations (such as DFT) which treat the hydrogen atom classically.
Acknowledgments. The MD simulations were made using the TBE branch of the questaal code (see questaal.org), which has been interfaced to the i-ipi suite by Michele Ceriotti. The movies were made using C++/vtk software built by Dimitar L. Pashov, using files generated for Michael Methfessel's xbs program.